PRESENTACIÓN DE CASOS
Distrofia corneal granular
Granular corneal dystrophy
Dra. Alexeide de la C. Castillo Pérez, I Dra. Daysi Vilches Lescaille, I Dr. Justo Luis Noriega, I Dr. Daneel Martínez Balido, II Dr. Bárbaro Ramón León Balbón,III Dra. Danysleidi León Bernal IV
I
Instituto Cubano de Oftalmología "Ramón Pando Ferrer". La
Habana, Cuba.
II Hospital General "Roberto Rodríguez".
Morón. Ciego de Ávila, Cuba.
III Policlínico "Manuel
Piti Fajardo". Villa Clara, Cuba.
IV Policlínico Centro. Villa Clara, Cuba.
RESUMEN
Las distrofias corneales constituyen un conjunto de enfermedades que presentan, en su mayoría, una baja incidencia y se caracterizan por acúmulo de material hialino o amiloide que disminuyen la transparencia corneal. La distrofia granular es una enfermedad autosómica dominante que presenta opacidades grises en el estroma superficial central de la córnea y se hacen visibles en la primera y segunda décadas de la vida, lo que provoca disminución de la visión más significativa cerca de los 40 años de edad. Presentamos dos casos clínicos de distrofia granular en pacientes hermanos de diferentes sexos, quienes acudieron a la consulta y refirieron visión nublada. El estudio de la historia familiar nos ayuda en el correcto diagnóstico y la biomicroscopia constituye el elemento más importante.
Palabras clave: distrofia corneal granular, material hialino, autosómica dominante.
ABSTRACT
Corneal dystrophies are a group of diseases that mostly have low incidence rates and are characterized by accumulation of hyaline or amyloid material that reduces the corneal transparency. Granular dystrophy is a dominant autosomal disease with gray opacities in the central superficial stroma of cornea, which are visible in the first and second decades of life and leads to significantly reduced vision when going into the 40 years of age. Here are two clinical cases of granular dystrophy in a pair of siblings who went to the doctor's because of blurred vision. The study of the family history helps the physician to reach a right diagnosis and the most important element is biomicroscopy.
Key words: granular corneal dystrophy, hyaline material, dominant autosomal disease.
INTRODUCCIÓN
Las distrofias corneales son enfermedades raras. Aun en una consulta especializada de córnea es infrecuente ver un paciente con esta enfermedad. Dentro de ellas las distrofias estromales tienen muy baja incidencia. El diagnóstico clínico es el más importante. La microscopia confocal y los estudios genéticos son exámenes valiosos que nos apoyan el criterio clínico.
La distrofia granular es de tipo estromal. En la biomicroscopia se observan depósitos en el estroma superficial central y en la membrana de Bowman. Es bilateral y asimétrica. El material encontrado en el estroma es de tipo queratohialino y puede ser producido por queratocitos anómalos. Por lo inusual de la afección, a continuación describimos las características de la enfermedad en dos hermanos diagnosticados en un Centro Oftalmológico guatemalteco.
PRESENTACIÓN DE CASOS
CASO 1
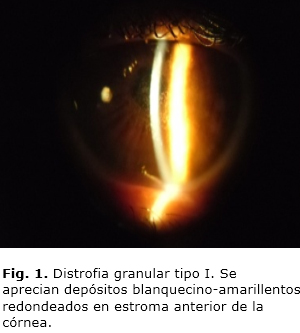
Paciente femenina, de 39 años de edad, con historia de disminución lenta y progresiva de la visión, quien acudió a consulta por visión nublada. En sus antecedentes patológicos familiares refirió que un hermano tenía "problemas visuales". Al examen biomicroscópico de ambos ojos se observaron depósitos blanco-amarillentos con aspecto de granos, en estroma anterior y medio, más numerosos en la zona central de la córnea y que dejan libre su periferia (Fig. 1). Al realizar examen de fondo de ojo no se encontraron alteraciones en el polo posterior. El ojo derecho (OD) tenía cuentadedos a 2 m y el ojo izquierdo (OI) a 3 m. Se le realizó refracción (OD –2,50 - 1,00 x 170, y el OI –3,00 - 1,25 x 140. La capacidad visual era de 0,1 y 0,3 para el OD y el OI, respectivamente. La queratometría en OD 44,00 x 160 y 43,00 x 10 y en el OI 44,50 x 150 y 42,00 x 140.
CASO 2
Paciente masculino, de 32 años de edad. Antecedente de visión nublada hace 4 años que mejoraba con el uso de lentes. Acudió a consulta para chequeo porque a su hermana se le diagnosticó una distrofia corneal y a la biomicroscopia presentaba depósitos blanco-amarillentos en estroma anterior y medio, más numerosos en la zona central de la córnea y que dejan libre su periferia con aspecto de granos (Fig. 2). No presentaba alteraciones del segmento posterior. El OD tenía cuentadedos a 3 m y el OI 0,1 de visión. En la refracción el OD –4,00 - 1,25 x 140 y el OI –3,50 - 2,00 x 120. La agudeza visual corregida OD 0,2 y OI 0,4. La queratometría OD 45,00 x 150 y 44,00 x 10 y el OI 44,00 x 10 y 42,00 x 120.
DISCUSIÓN
Las distrofias corneales son un grupo de alteraciones hereditarias caracterizadas por depósitos anormales de material amiloide, hialino o mixto en la córnea, que ocasionan pérdida de la transparencia corneal. El diagnóstico de la distrofia se plantea cuando esta se presenta en edades en que se presupone su aparición, las características de bilateralidad, progresión en el tiempo, localización central en la córnea y ausencia de inflamación.1
En
este estudio presentamos a dos hermanos. La primera paciente descrita acudió
a consulta por presentar visión nublada y posteriormente fue citado su
hermano para chequeo porque tenía historia de cambio frecuente de lentes
para tratamiento de su problema visual. Otros familiares no estuvieron disponibles
para la valoración de las lesiones corneales con el fin de poder establecer
si existía variación clínica intrafamiliar. Las características
de los depósitos corneales encontrados en ambos hermanos fueron similares.
Se diagnosticó distrofia corneal
granular tipo I o clásica, conocida también como distrofia de
Groenouw I, distrofia en miga de pan y distrofia anular de Bücklers, porque
las características biomicroscópicas de la córnea son típicas
de esta enfermedad.
Visión borrosa, deslumbramiento y mayor dificultad para la visión
de noche son los síntomas más frecuentes. A medida que progresa
se produce disminución de la agudeza visual. Puede observarse desde la
primera década, aunque las alteraciones visuales pueden presentarse después
de la cuarta década. Un tercio de los pacientes presentan erosión
corneal recidivante. Se transmite con carácter autosómico dominante.
Estudios en sujetos de diversos orígenes étnicos han demostrado
que este tipo de distrofia es causada casi exclusivamente por la sustitución
de arginina a triptófano en el aminoácido 555 (R555W) de TGFBI.2,3
Otros tipos de distrofia granular se descartaron, como la de Avellino o tipo II, ya que en ella se observan opacidades más gruesas, con morfología de anillos, discos o copos de nieve, pocas en número, que no fueron evidentes en nuestros pacientes, y no están asociadas generalmente a erosiones recurrentes. Se presenta generalmente en la segunda década de la vida. Su progresión es lenta.4 El tipo III se presenta en la infancia. Los depósitos se localizan más superficialmente en la córnea. Son frecuentes las erosiones recurrentes. La capa de Bowman se encuentra destruida. Puede encontrarse asociada a distrofia reticular, queratocono y distrofia de conos de la retina.5 Debemos hacer el diagnóstico diferencial con otras distrofias estromales como la reticular y la macular. En la distrofia reticular se observan líneas grises en el estroma que dan el aspecto de vidrio esmerilado. La distrofia macular es una enfermedad autosómica recesiva y en ella observan manchas blanco-grisáceas con bordes indefinidos. El material encontrado es mucopolisacáridos.1-6
La distrofia central de Schnyder es otra distrofia estromal que se presenta en la primera década de la vida con opacidades redondas, anulares u ovales, compuestas de finos cristales policromáticos que contienen colesterol. Rara vez se afecta la agudeza visual. Las distrofias corneales epiteliales y endoteliales se descartan por su diferente localización en la córnea. El tratamiento quirúrgico debe decidirse en cada caso en dependencia de la visión y la sintomatología por erosiones recurrentes. La queratectomía con láser de excímero (PTK) se puede utilizar en casos donde predominan los depósitos superficiale.7-9 La queratoplastia penetrante ha sido el tratamiento clásico. Los resultados con esta cirugía son buenos, pero se presenta frecuentemente recidiva en el injerto.10
Las distrofias corneales constituyen un grupo de enfermedades relativamente poco frecuentes y dentro de ellas la distrofia granular no es una excepción. Su bilateralidad, localización y edad de aparición son típicas. El estudio de la historia familiar nos ayuda en el correcto diagnóstico, y la biomicroscopia constituye el elemento más importante. Los avances en la genética nos conducen por el camino que nos enriquecerá en su conocimiento y tratamiento.
REFERENCIAS BIBLIOGRÁFICAS
1. Barraquer RI, Toledo MC, Torres E. Distrofia y degeneraciones corneales. Atlas y texto. Barcelona: Espaxs S.A; 2004.
2. Castelo Branco B, Chalita MR, Casanova FH, Branco AB, Allemann N, Freitas D. Amorphous corneal dystrophy; ultrasound biomicroscopy findings in two cases. Cornea. 2002;21(2):220-2.
3. Olivera LA de. Distrofia corneana amorfa posterior: relato de caso. Arg Bras. Oftalmol. 2006;69(6):945-7.
4. Donate López J, Iradier Urrutia MT, Bohórquez Rodríguez P, Castillo Gómez A. Distrofia polimorfa posterior. A propósito de 3 casos. Studium ophthalmologicum. 2000 [citado 12 de enero de 2015];XIX(1). Disponible en: http://www.oftalmo.com/studium/studium2000/stud00-1/00a07.htm
5. Arffa R. Enfermedades de la córnea. Barcelona: Harcourt Brace; 2000.
6. Cremona G. Distrofias corneales. En: Chiaradía P. La córnea en apuros. Buenos Aires: Científicos-Argentinas; 2006:110-21.
7. Sutton GL, Kim P. Laser in situ Keratomleusis in 2010-a review. Clin Experiment Ophthalmol. 2010;38(2):192-210.
8. Nettune GR, Pflugfelder SC. Post-Lasik tear dysfunction and dysestesia. Ocul Surf. 2010;8(3):135-45.
9. Farid M, Garg S, Steinert RF. Fentosecond Laser-assisted penetrating keratoplasty. Holanda: Cornea Surgery. 2011:1349-55.
10. Van Meter W, Katz DG. Keratoplasty suturing techniques. Cornea. 2005;123:1481-90.
Recibido:
23 de febrero de 2014.
Aprobado: 3 de marzo de 2015.
Dra. Alexeide de la C. Castillo Pérez. Instituto Cubano de Oftalmología "Ramón Pando Ferrer". Ave. 76 No. 3104 entre 31 y 41 Marianao, La Habana, Cuba. Correo electrónico: alexcastillo@infomed.sld.cu